The Institute for Evolution and Biodiversity (IEB) currently integrates six groups working on evolutionary ecology of animals, plants and microorganisms, phylogeny and evolution, aquatic ecology, biocomplexity, and evolutionary bioinformatics.
Our core question is how biodiversity and biocomplexity at all levels of the biological hierarchy arises through evolutionary processes.
1) Which are the factors that contribute to competition success of invading species?
Invertebrates: Three sympatric species of Gammaridae (Amphipoda, Crustacea) show a specific spatial distribution in the upper catchment of the River Lippe, eastern Northrhine-Westphalia, Germany. Stretches of the streams are characterized by temporal flow intermittency, which originates from the particular hydrogeologic situation (karst streams).Gammarus fossarum lives in the upstream reaches, whereas Gammarus pulex prefers the mid-stream, temporary sections. Aside from these native species, Echinogammarus berilloni, a neozoon, is currently invading the stream system from the downstream main stem. Obviously, this species is going to displace Gammarus pulex by its upstream conquest.In order to understand the patterns and dynamics of the modern regional biodiversity, the underlying mechanisms are studied in comparative field and laboratory experiments on this examplary system amongst others. Here we test the ecological valences and life traits with respect to abiotic factors, food preferences, and effect of stressors such as hydrologic perturbation and the unpredictability of environmental factors on the performance of the species. By studying the metapopulation structures we further will trace the dispersial history of the species.
Limnology Group - [ Prof. Dr. Elisabeth Meyer ]
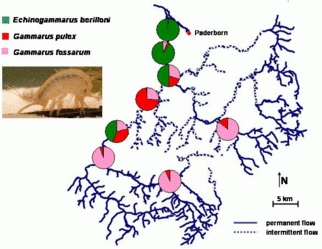
© IEB - Limnology Group 2) Coevolution between the red flour beetle and Bacillus thuringiensis bacteria
Host-parasite interactions are ideal systems for studying fast evolutionary processes. We use the red flour beetle Tribolium castaneum as a host and Bacillus thuringiensis (Bt) as a specific micro-parasite to track co-evolutionary processes in the laboratory. We then analyse the genomic underpinnings of such reciprocal adaptation using next-generation sequencing technology.
This is part of the research of the Animal Evolutionary Ecology Group - [ Prof. Dr. Joachim Kurtz ]
Find out more about the principles of animal evolution on their research page.
The picture shows larvae of the red flour beetle eating on a dead conspecific that was killed from an infection with B. thuringiensis. It highlights cannibalism as a possible natural route of transmission of B. thuringiensis.© IEB - Momir Futo 3) Evolvability, epistasis and molecular robustness
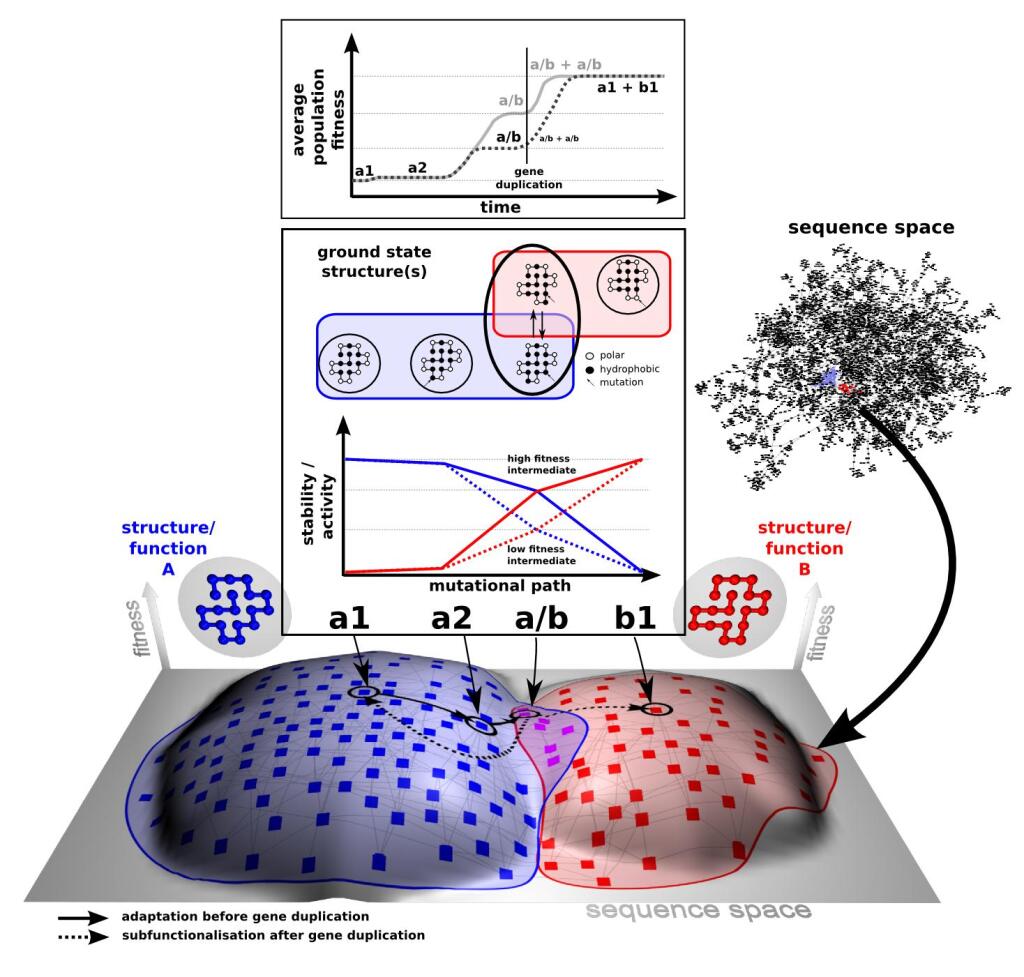
The key to understanding adaptability of a given protein is to determine the shape of fitness landscapes of both 'old' (blue) and 'new' (red) functions in relation to the accessible sequence space (panel C). By definition ancestral proteins that have descendants with different primary functions are evolvable, i.e. they should score relatively high on the evolvability scale (panel A). Therefore resurrecting these ancestral proteins (internal nodes indicated with circles in panel A), based on the evolutionary history inferred from related present day proteins (indicated with squares in panel A), should provide insight into which biophysical/biochemical properties are important for 'evolvability’. Furthermore these resurrected ancestors could be starting points for alternative evolutionary paths.
Molecular Evolution and Bioinformatics Group - [ Prof. Dr. Erich Bornberg-Bauer ]
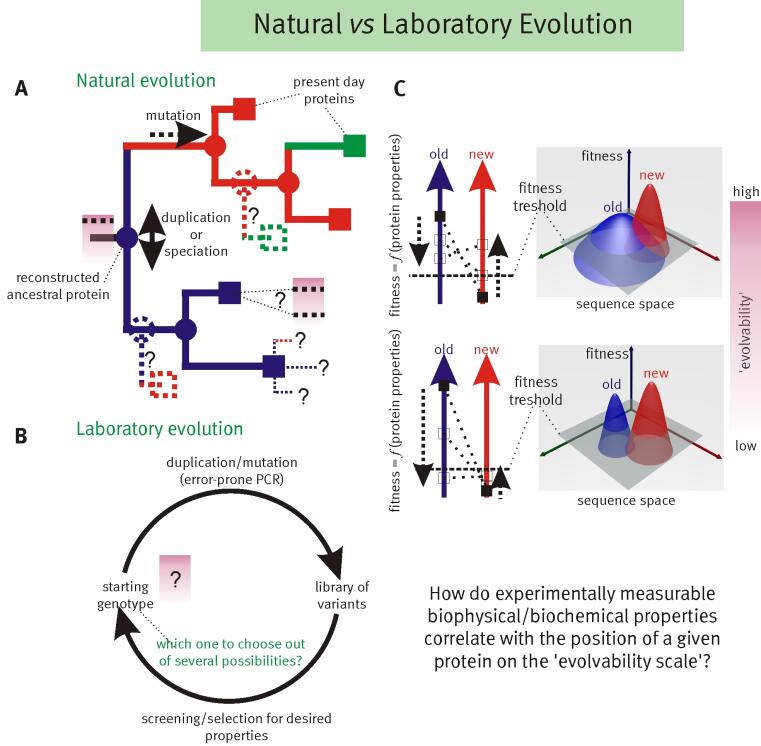
Adaptation of proteins to new function: using the evolutionary history of proteins to help understand their evolvability© IEB Molecular Evolution and Bioinformatics 4) Modular evolution of genes, proteins and networks
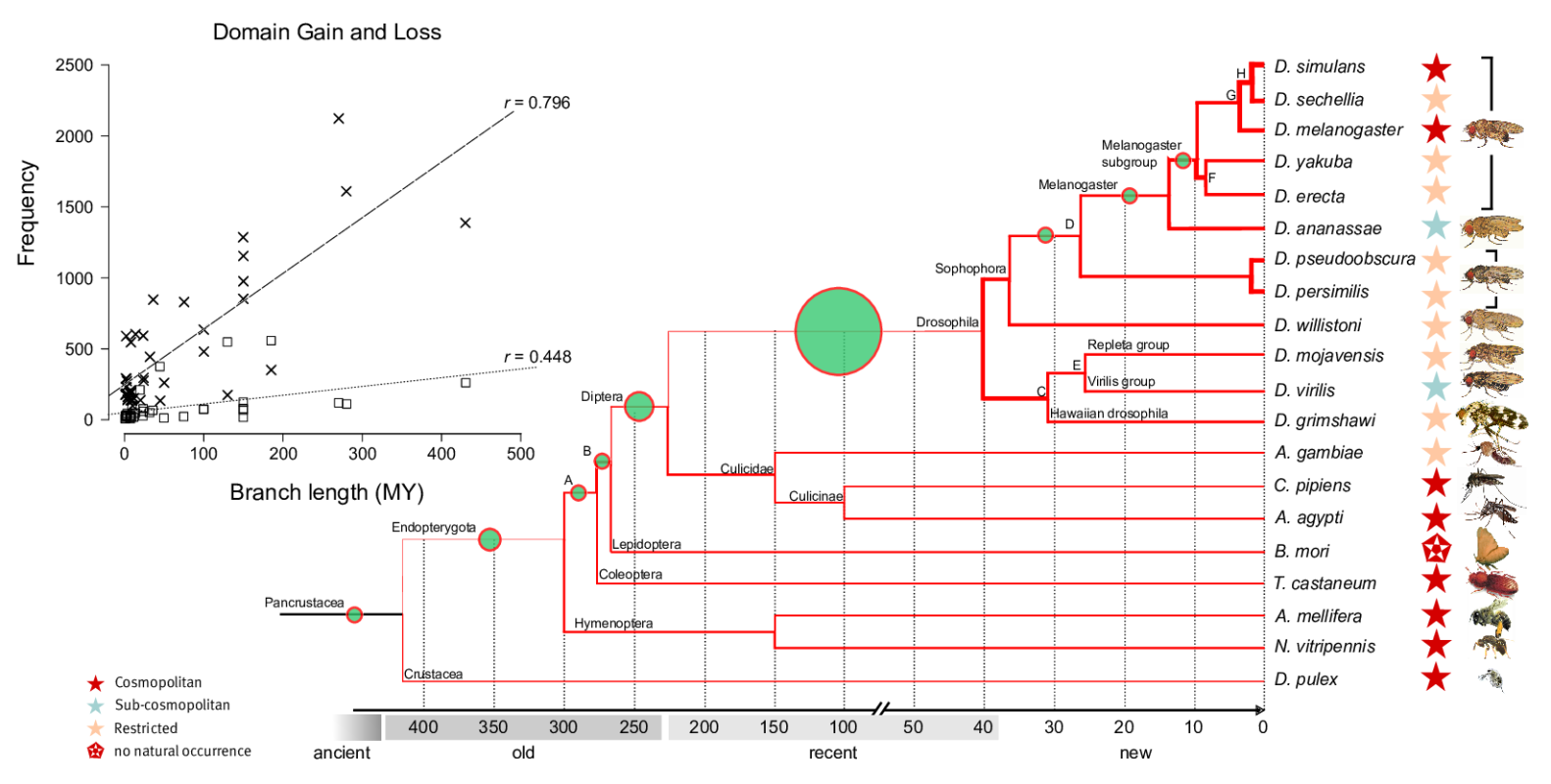
Dynamics and adaptive benefits of domain rearrangementsModularity is a hallmark of molecular evolution. Whether considering gene regulation, the components of metabolic pathways or signalling cascades -- the ability to reuse autonomous modules in different molecular contexts can expedite evolutionary innovation. Over times scales of several 100 Mys, the evolution of protein coding genes is dominated by the modular rearrangements of protein domains, their evolutionary, structural and functional units. While a small core of arrangements is universal, a large fraction of multi-domain arrangements is species specific and has been created recently via gene duplication, fusion and terminal loss of domains. Surprisingly, thousands of domains are completely (i.e. all copies within a genome) lost from genomes along every lineage in a stochastic manner, i.e. at a fairly constant rate of ca. four domains per million years.
Novel domains are rarely fixed and arise either as their own genes or terminally, by extension of existing reading frames. Novel domains are under strong selection pressure and confer a strong fitness value as they rapidly attain high copy numbers within the genomes and are involved in biotic defence, reproduction and development.
Using cross-species genomic comparisons and population genomics we investigate the genetic mechanisms and biophysical constraints of domain emergence and develop algorithms for rapid screening of many genomes, understanding their phylogenies and comparing, aligning and clustering sequences.Molecular Evolution and Bioinformatics Group - [ Prof. Dr. Erich Bornberg-Bauer ]
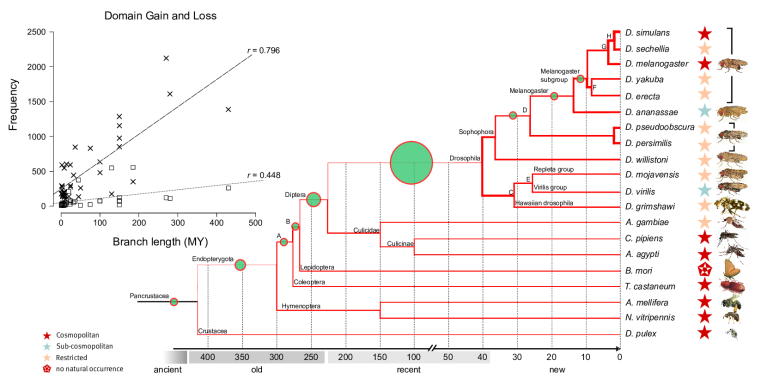
Domain loss (steep line in insert) and gain (flatter line) across insect genomes. High rates (thick lines in tree) of loss are contrasted by low rates of gain (green circles). 
P-value transformed TermLogo of functional groups with emerging domains in the insect clade (as of 2010) indicate that most novel domains confered functional benefits in the context of environmental adaptation and development.© IEB - Molecular Evolution & Bioinformatics Group 5) Evolutionary origin, fixation and functions of de novo protein coding genes
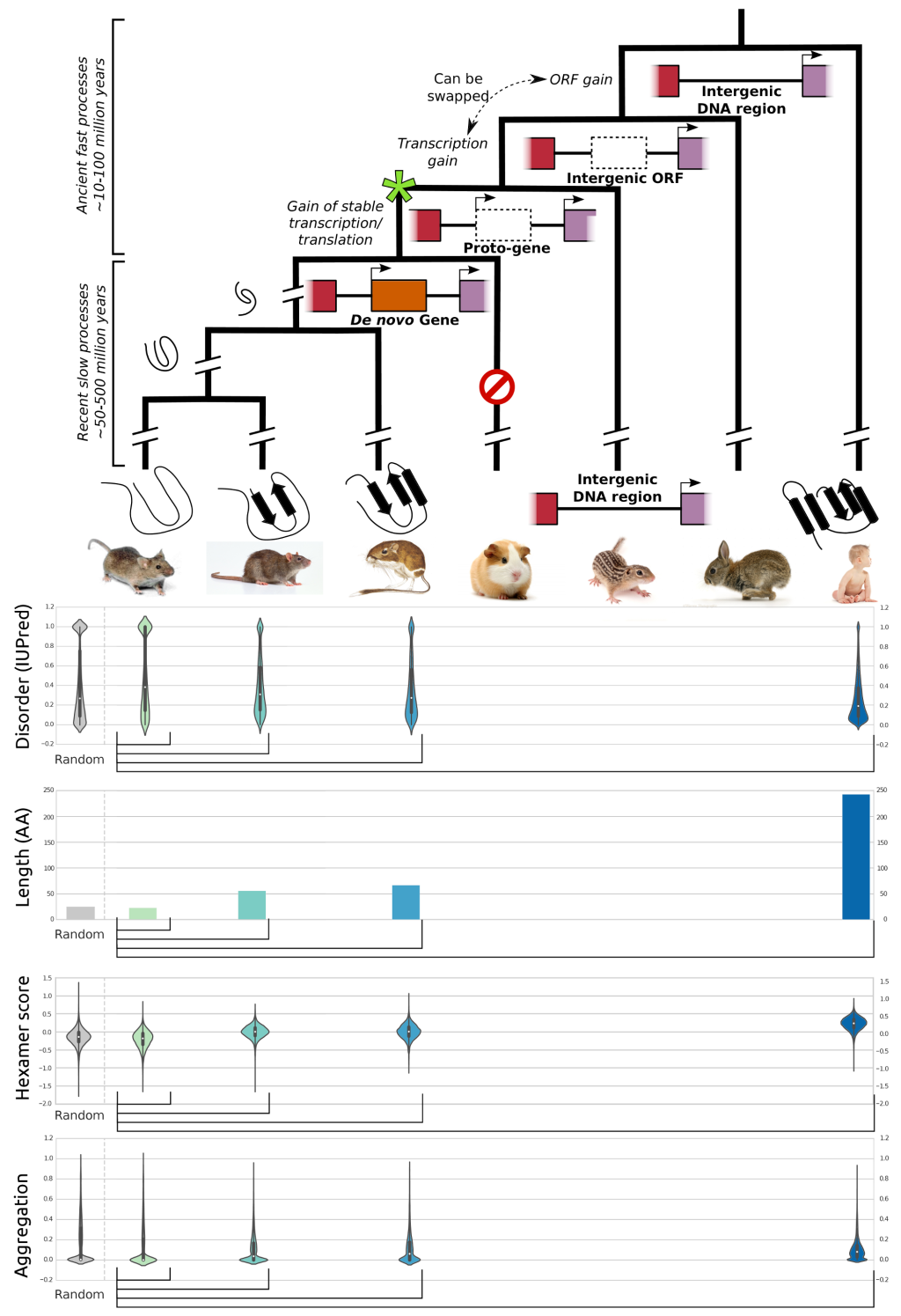
With every new genome sequenced a couple of hundred proposed genes remain ''orphans'' because computational methods could not assign any orthologues, even to closely related and well annotated species. Presumably many of these (lineage-specific) genes are transcribed, sometimes translated and proteins functional and adaptive, at least under some (possibly unknown) conditions. De novo emergence is not only against current believe that most novel genes emerge from old ones, it is also difficult to reconcile with a biophysical perspective because novel reading frames emerging from previously non-coding matter must be considered extremely unlikely: they would most likely be disordered, aggregate and thus be deleterious or, at least be purged for purely energetic reasons. So, where do new coding genes actually come from, how do they function and how is their -- potentially detrimental -- expression regulated?
We ask where novel protein coding genes come from and how genomic novelties and rearrangements trigger adaptation and spur developmental transitions. Using comparative genomics and biophysical analyses (computational and experimental) we test their properties and functions. We found that most genetic novelty comes from novel domains but also many completely new reading frames emerge, e.g. across the insect tree, with an estimated frequency of 500 new genes in the wake of each speciation event. This former process has been termed ''grow slow and moult'' because some novel domains later lose their initially stabilizing parent protein and become independent and amenable for further rearrangements.
Molecular Evolution and Bioinformatics - [ Prof. Dr. Erich Bornberg-Bauer ]
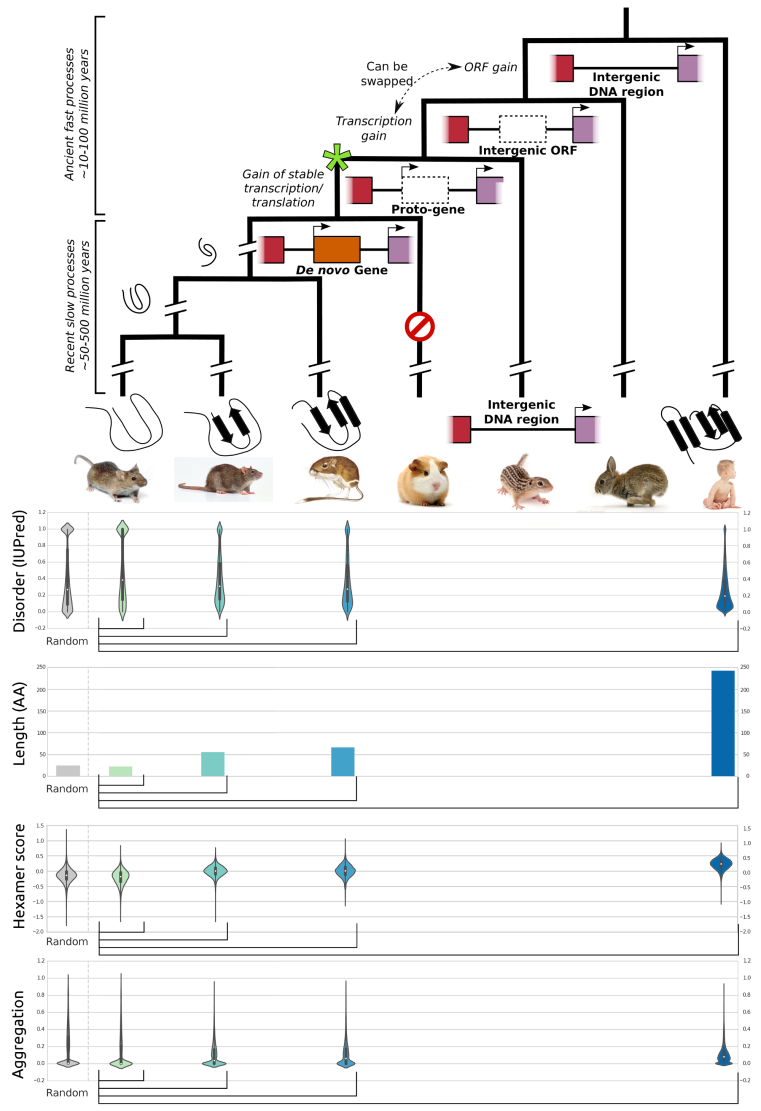
Schematic depiction of de novo gene emergence. Sequence properties of mouse ORFs of various age classes and randomly generated ORFs are shown underneath the tree. Conservation levels of ORF classes are shown by brackets indicating the most distant outgroup with detectable sequence similarity.© IEB - Molecular Evolution & Bioinformatics Group 6) Evolutionary and functional genomics of (mostly social) insects
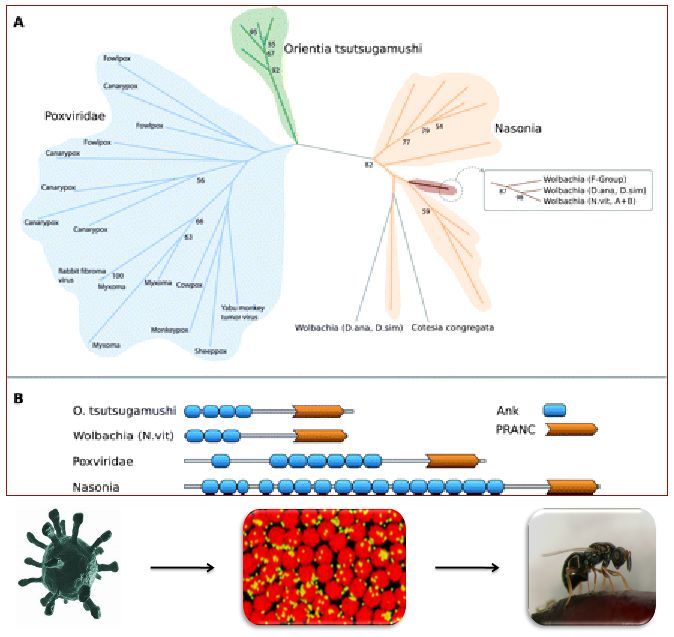
Sociality is considered as one of the major transitions in evolution but only little of the underlying genomic basis and the associated selectable traits are known. Social insect are an excellent study objects because their genomes are relatively simple to analyse and many speciation events gave rise to morphologically and ecologically diverse species within a relatively short time period. Furthermore, in insects, sociality has independently evolved at least twice, in hymenoptera (comprising ants and bees) and in termites. Both groups also show a striking reversal of the otherwise widely spread trade-off between longevity and fecundity. Furthermore, loss and/or reversal of social behaviour has been observed since several ants parasitize or enslave the colonies of evolutionary very closely related species.
We have investigated genomes and transcriptomes of several insects, most of which are either social or closely related outgroups thereof. Using standard bioinformatics techniques and several of our in-house algorithms we could identify horizontally transferred genes from bacterial parasites, several genes under adaptation and novel genes which were instrumental for the ecological success of social insects. In comparing social with non-social insects, we found that both, novel genes and rewiring of regulatory networks, play a big role for the regulation of sociality. We also consider the epigenetic marks and effects from methylation/acetylation and of small regulatory RNAs on the differentiation of individuals during their development to test the possible role of epigenetic marks in general and its effect on de novo genes and on rearranged genes.
Molecular Evolution and Bioinformatics Group - [ Prof. Dr. Erich Bornberg-Bauer ]
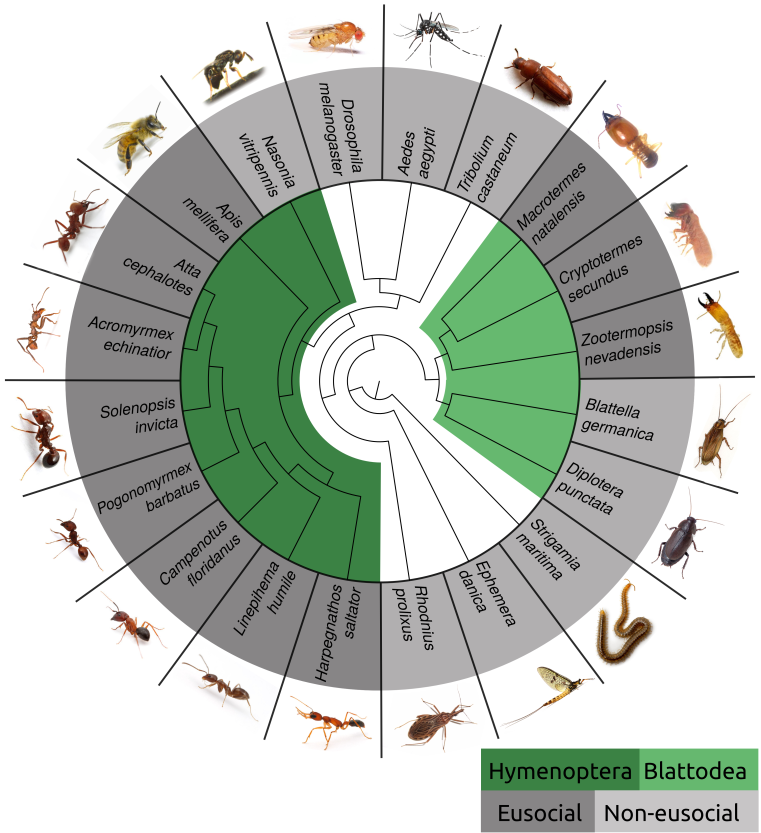
Eusociality has independently evolved in the insect clades Hymenoptera (bees, wasps, ants, bumble bees) and Blattodea (cockroaches and termites). Comparative genomics of eusocial insects and closely related non-eusocial species can shed light on the evolution of eusociality© IEB Molecular Evolution and Bioinformatics 8) Evolutionary (ecology) theory
A classical branch of evolutionary theory, population genetics, is concerned with the study of allele frequency changes in a population. Quantities like the invasion probability of a new genetic variant, an allele, the fixation or extinction time of an allele and the allele frequency distribution within a population are at the core of this research field. In our research, we analytically compute or numerically simulate these quantities to predict the evolutionary outcome in different biological scenarios. There are two recurring themes in our research: (i) The analysis of stable polymorphisms in different evolutionary scenarios, e.g. self-incompatibility alleles in plants or funghi. (ii) The study of effects of ecological processes like species interactions, environmental change or spatial structure on the evolutionary dynamics. For example how does genotype-specific dispersal, e.g. habitat choice, affect the adaptive potential in a changing, spatially structured environment?
Peter Czuppon
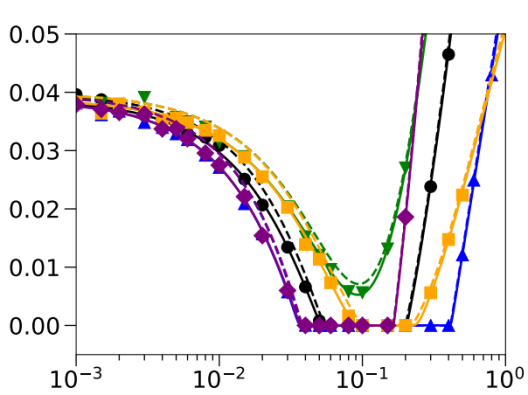
The adaptation probability (y-axis) of a population to a habitat that undergoes environmental change when the dispersal rate is varied (x-axis). Different colors correspond to different genotype-dependent dispersal schemes. Solid and dashed lines are different theoretical predictions, symbols correspond to individual-based stochastic simulation results.© acoban